Comment faire fonctionner IGV en utilisant la communication par socket, et l'histoire de la création d'un Ruby Gem en utilisant cette méthode
introduction
IGV est un logiciel appelé Genome Browser. Vous pouvez parcourir différents fichiers tels que le fichier bam de la lecture alignée, le fichier lit dans lequel les informations de position sur le génome sont annotées et le fichier gff3 dans lequel les informations génétiques sont écrites. De nombreuses personnes impliquées dans la bioinformatique connaissent ce logiciel.

- L'image provient de la page d'accueil IGV
Bien qu'il s'agisse d'un tel IGV, il peut prendre un certain temps pour le faire fonctionner en cliquant avec la souris.
- Précisez le génome de référence
- Chargez les fichiers requis
- Regardez autour de vous les endroits auxquels vous faites attention un par un
- Prenez une capture d'écran et enregistrez-la en tant qu'image
C'est amusant de vérifier visuellement les résultats, et je pense que c'est une tâche très importante, mais d'un autre côté, je veux aussi automatiser autant que possible les tâches de routine. Par conséquent, envisagez d'utiliser l'IGV à partir d'un langage de programmation.
- Récemment, il semble que igv.js réalisé avec JavaScript a été activement développé et largement utilisé. .. Ceci n'est pas couvert dans cet article en raison d'une recherche insuffisante. (Mais j'aimerais finalement faire un article sur Qiita.)
Faites fonctionner l'IGV en utilisant la communication par socket
En fait, IGV peut être exploité à partir du port 60151 en utilisant la communication par socket.
Il y a un onglet appelé avancé dans Affichage> Préférences dans la barre de menus. Si Activer le port n'est pas coché, cochez ici.
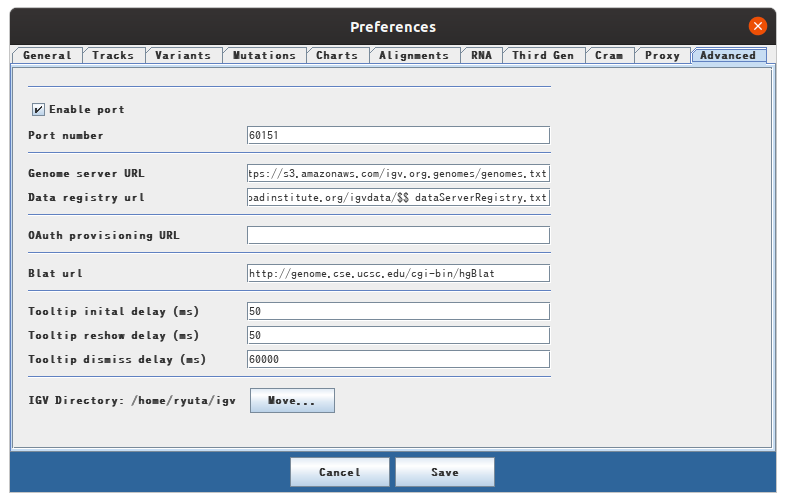
Vous pourrez alors utiliser le port 60151 pour diriger l'IGV en utilisant la communication par socket.
Liste des commandes pouvant être utilisées pour faire fonctionner IGV
J'ai traduit la référence officielle en japonais en utilisant la traduction DeepL.
| Command | Description |
|---|---|
| new | Créez une nouvelle session. Supprimez toutes les pistes à l'exception de l'annotation du génome par défaut. |
| load file | Lire des données ou des fichiers de session. Spécifiez le chemin d'accès complet ou l'URL séparés par des virgules. |
| collapse trackName | Réduit le trackName spécifié. Si vous ne spécifiez pas trackName, toutes les pistes seront réduites. |
| echo | En réponse"echo "Retour.(pour le test) |
| exit | Quittez l'application IGV. |
| expand trackName | Développe le trackName spécifié. Si trackName n'est pas spécifié, toutes les pistes seront développées. |
| genome genomeIdOrPath | Sélectionnez un génome par identifiant ou chargez le génome (indexé fasta) à partir du chemin spécifié. |
| goto locus or listOfLoci | Faites défiler jusqu'à un seul locus ou une liste de locus séparés par des espaces. Si une liste est fournie, ces positions d'assise sont affichées dans une vue en écran partagé. Toute syntaxe valide dans la zone de recherche IGV convient. |
| goto all | Faites défiler jusqu'à la vue du génome entier. |
| group option | Piste d'alignement uniquement. Groupez les alignements avec l'une des options suivantes: BRIN, SAMPLE, READ_GROUP, LIBRARY, FIRST_OF_PAIR_STRAND, TAG, PAIR_ORIENTATION, MATE_CHROMOSOME, SUPPLEMENTARY, MOVIE, ZMW, HAPLOTYPE, READ_ORDER, NONE, BASE_AT_POS |
| region chr start end | Définissez une région d'intérêt entourée de deux loci (par exemple, région chr1 100 200). |
| maxPanelHeight height | Définit le nombre de pixels verticaux (hauteur) pour chaque panneau inclus dans l'image. Les images créées à partir de commandes de port ou de scripts batch ne sont pas limitées aux données affichées à l'écran. En d'autres termes, vous pouvez inclure le panneau entier dans votre image, pas seulement ce qui est affiché dans la zone d'écran déroulante. La valeur par défaut de ce paramètre est 1000, augmentez cette valeur pour afficher plus de données ou diminuez cette valeur pour créer des images plus petites. |
| setLogScale(true or false) | |
| setSleepInterval ms | Définit le temps de retard (veille) en millisecondes. L'intervalle de sommeil est appelé entre des commandes consécutives. |
| snapshotDirectory path | Définissez le répertoire pour écrire l'image. |
| snapshot filename | Enregistrez un instantané de la fenêtre IGV dans un fichier image. Si le nom de fichier est omis, un fichier PNG avec le nom de fichier généré en fonction de la trajectoire est écrit. Si le nom de fichier est spécifié, l'extension du nom de fichier détermine le format du fichier image..png、.jpg, ou.Doit être svg. |
| sort option locus | Trier les pistes avec des numéros de copie alignés ou segmentés. La valeur appliquée à l'option de numéro de copie segmenté est(1)AMPLIFICATION et SUPPRESSION, numéros de copie segmentés,(2)L'alignement suit POSITION, STRAND, BASE, QUALITY, SAMPLE, READGROUP, INSERSTSIZE, FIRSTOFPAIRSTRAND, MATECHR, READORDER et READNAME. L'option est insensible à la casse. Vous pouvez spécifier un lieu pour définir une seule position ou plage. Si vous ne spécifiez pas d'option, le tri est effectué en fonction de la zone affichée ou de la position centrale de la zone affichée. |
| squish trackName | Écrasez le trackName donné. trackName est facultatif et toutes les pistes d'annotation seront écrasées si elles ne sont pas spécifiées. |
| viewaspairs trackName | Mode d'affichage de la piste d'alignement"View as pairs "Mis à. |
| preference key value | Définit temporairement le paramètre nommé key sur la valeur spécifiée. Ce paramètre n'est valide que jusqu'à ce que l'IGV soit arrêté. |
Manipuler IGV à partir du langage de programmation
Java
Comme IGV est un logiciel développé en Java, des exemples officiels sont également écrits en Java.
Socket socket = new Socket("127.0.0.1", 60151);
PrintWriter out = new PrintWriter(socket.getOutputStream(), true);
BufferedReader in = new BufferedReader(new InputStreamReader(socket.getInputStream()));
out.println("load na12788.bam,n12788.tdf");
String response = in.readLine();
System.out.println(response);
out.println("genome hg18");
response = in.readLine();
System.out.println(response);
out.println("goto chr1:65,827,301");
//out.println("goto chr1:65,839,697");
response = in.readLine();
System.out.println(response);
out.println("snapshotDirectory /screenshots");
response = in.readLine();
System.out.println(response);
out.println("snapshot");
response = in.readLine();
System.out.println(response);
R
Maintenant, qu'en est-il des langages de programmation autres que Java? Si vous utilisez R, je ne suis pas très familier avec celui-ci, mais vous pouvez l'utiliser car une bibliothèque pour faire fonctionner IGV sera fournie à partir du bioconducteur. Une petite recherche va frapper le logiciel igvR. Cela semble utiliser igv.js, donc il peut ne pas faire fonctionner automatiquement l'IGV sur le bureau ...
Python
C'est un script un peu ancien, mais igv.py créé par Brent Pedersen, qui a récemment développé énergiquement des outils bioinformatiques utilisant le langage Nim. -Il existe un outil appelé terrain de jeu / blob / master / igv / igv.py). Il s'agit d'une petite bibliothèque qui encapsule la communication de socket ci-dessus.
Ruby
J'aime Ruby. ** Nani Nani? Y a-t-il peu d'outils? Ensuite, vous pouvez le fabriquer vous-même! ** Donc, en me référant au script de Brent Pedersen ci-dessus, j'ai créé un outil appelé ruby-igv qui peut faire fonctionner IGV à partir du langage Ruby. Maintenant, vous pouvez facilement utiliser IGV à partir du langage Ruby.
https://github.com/kojix2/ruby-igv
L'utilisation est comme ça.
igv = IGV.new
igv.load 'na12788.bam'
igv.genome 'hg18'
igv.go 'goto chr1:65,827,301'
igv.save 'image.png'
C'est toujours un outil fraîchement fabriqué. Si vous rencontrez des problèmes, des bogues ou des demandes, veuillez les signaler à issue sur Github. Bien entendu, les pull requests sont également les bienvenues.
en conclusion
Non limité à la bioinformatique, l'attention tend à se concentrer sur la manière de combiner les outils existants pour atteindre l'objectif et la maîtrise des outils. Et lorsque vous créez un outil, vous pouvez penser que le but et les moyens sont inversés. Mais dans un sens, je pense que c'est une façon de penser très égoïste et bornée. Plus il y a de gens qui créent et publient plus d'outils, plus le monde sera pratique, en expansion et prospère. Créons et publions des outils plus librement. (Parce que ça ne doit pas être Ruby)
Recommended Posts