How to operate IGV using socket communication, and the story of making a Ruby Gem using that method
Introduction
IGV is software called a genome browser. You can browse various files such as the bam file of the aligned read, the bed file in which the position information on the genome is annotated, and the gff3 file in which the genetic information is written. If you have been involved in bioinformatics, you probably know this software.

- The image is from IGV homepage
Although it is such an IGV, it may take some time to operate it while clicking with the mouse.
- Specify the reference genome
- Load the required file
- Look around the places you are paying attention to one by one
- Take a screenshot and save it as an image
It's fun to check the results visually, and I think it's a very important task, but on the other hand, I also want to automate routine tasks as much as possible. Therefore, consider operating the IGV from a programming language.
- Recently, it seems that igv.js made with JavaScript has been actively developed and widely used. .. This is not covered in this article due to insufficient research. (But eventually I'd like to make a Qiita article.)
Operate IGV using socket communication
In fact, IGV can be operated from port 60151 using socket communication.
There is a tab called advanced in View> Preference on the menu bar. If Enable port is not checked, check here.
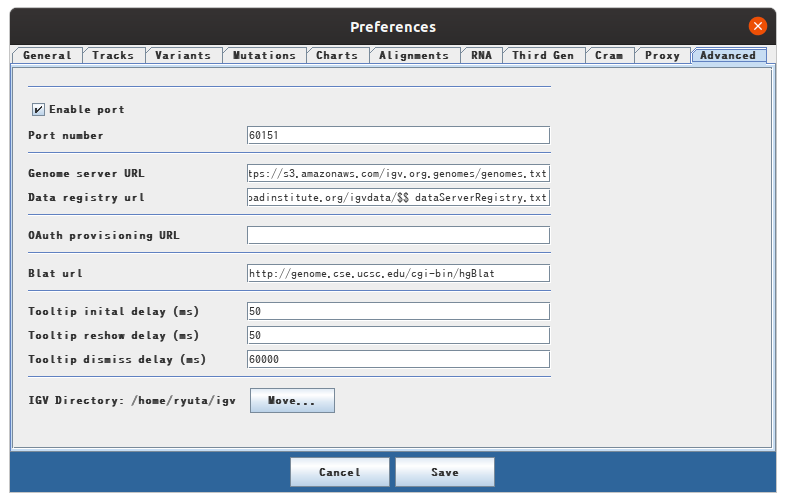
You will then be able to use port 60151 to steer the IGV using socket communication.
List of commands that can be used to operate IGV
I translated the official reference into Japanese using DeepL Translator.
| Command | Description |
|---|---|
| new | Create a new session. Remove all tracks except the default genome annotation. |
| load file | Read data or session files. Specify the full path or URL separated by commas. |
| collapse trackName | Collapses the specified trackName. If you do not specify trackName, all tracks will be collapsed. |
| echo | In response"echo "Returns.(for test) |
| exit | Quit the IGV application. |
| expand trackName | Expands the specified trackName. If trackName is not specified, all tracks will be expanded. |
| genome genomeIdOrPath | Select the genome by id or load the genome (indexed fasta) from the specified path. |
| goto locus or listOfLoci | Scroll to a single locus or a space-separated list of loci. If a list is provided, these sitting positions are displayed in split screen view. Any syntax that is valid in the IGV search box is fine. |
| goto all | Scroll to View the entire genome. |
| group option | Alignment track only. Group the alignments with one of the following options: STRAND, SAMPLE, READ_GROUP, LIBRARY, FIRST_OF_PAIR_STRAND, TAG, PAIR_ORIENTATION, MATE_CHROMOSOME, SUPPLEMENTARY, MOVIE, ZMW, HAPLOTYPE, READ_ORDER, NONE, BASE_AT_POS |
| region chr start end | Define a region of interest surrounded by two loci (for example, region chr1 100 200). |
| maxPanelHeight height | Sets the number of pixels (height) in the vertical direction for each panel included in the image. Images created from port commands or batch scripts are not limited to the data displayed on the screen. In other words, you can include the entire panel in your image, not just what is displayed in the scrollable screen area. The default value for this setting is 1000, increase this value to display more data and decrease this value to create smaller images. |
| setLogScale(true or false) | |
| setSleepInterval ms | Sets the delay (sleep) time in milliseconds. The sleep interval is called between consecutive commands. |
| snapshotDirectory path | Set the directory to write the image. |
| snapshot filename | Save a snapshot of the IGV window to an image file. If filename is omitted, a PNG file with the filename generated based on the trajectory will be written. If filename is specified, the filename extension determines the format of the image file..png、.jpg, or.Must be svg. |
| sort option locus | Sort tracks with aligned or segmented copy numbers. The value applied to the segmented copy number option is(1)AMPLIFICATION and DELETION of segmented copy numbers,(2)POSITION, STRAND, BASE, QUALITY, SAMPLE, READGROUP, INSERSTSIZE, FIRSTOFPAIRSTRAND, MATECHR, READORDER, and READNAME of the alignment track. option is case insensitive. You can specify locus to define a single position or range. If you do not specify an option, the sort is performed based on the displayed area or the center position of the displayed area. |
| squish trackName | Squish the given trackName. trackName is optional and if not specified, all annotation tracks will be squished. |
| viewaspairs trackName | Alignment track display mode"View as pairs "Set to. |
| preference key value | Temporarily sets the setting named key to the specified value. This setting is valid only until the IGV is shut down. |
Manipulate IGV from a programming language
Java
Since IGV is software developed in Java, the official example is also written in Java.
Socket socket = new Socket("127.0.0.1", 60151);
PrintWriter out = new PrintWriter(socket.getOutputStream(), true);
BufferedReader in = new BufferedReader(new InputStreamReader(socket.getInputStream()));
out.println("load na12788.bam,n12788.tdf");
String response = in.readLine();
System.out.println(response);
out.println("genome hg18");
response = in.readLine();
System.out.println(response);
out.println("goto chr1:65,827,301");
//out.println("goto chr1:65,839,697");
response = in.readLine();
System.out.println(response);
out.println("snapshotDirectory /screenshots");
response = in.readLine();
System.out.println(response);
out.println("snapshot");
response = in.readLine();
System.out.println(response);
R
Now, what about programming languages other than Java? If you are using R, I'm not very familiar with it, but you can use it because a library for operating IGV will be provided from around bioconductor. A little search will hit the software igvR. This seems to utilize igv.js, so it may not automatically operate the IGV on the desktop ...
Python
It's a bit old script, but igv.py created by Brent Pedersen, who has recently been energetically developing bioinformatics tools using the Nim language. -There is a tool called playground / blob / master / igv / igv.py). This is a small library that wraps the socket communication above.
Ruby
I like Ruby. ** Nani Nani? Are there few tools? Then you can make it yourself !** So, referring to Brent Pedersen's script above, I created a tool called ruby-igv that can operate IGV from Ruby language. Now you can easily operate IGV from the Ruby language.
https://github.com/kojix2/ruby-igv
The usage is like this.
igv = IGV.new
igv.load 'na12788.bam'
igv.genome 'hg18'
igv.go 'goto chr1:65,827,301'
igv.save 'image.png'
It's still a freshly made tool. If you encounter a rough cut, a bug, or find a request, please report it to issue on Github. Of course, pull requests are also welcome.
in conclusion
Not limited to bioinformatics, attention tends to be focused on how to combine existing tools to achieve the purpose and how to master the tools. And when you are making a tool, you may think that the purpose and means are reversed. But in a sense, I think it's a very selfish and narrow-minded way of thinking. The more people who make and publish tools, the more convenient, expanding and prosperous the world will be. Let's make and publish tools more freely. (Because it doesn't have to be Ruby)
Recommended Posts