2 . Il existe deux types de données de recherche d'échantillons pour ChemTHEATER, les données mesurées et les échantillons, mais cette fois, nous utiliserons les deux.
Le manuel d'utilisation de ChemTHEATER peut être obtenu sur le site Wiki ChemTHEATER .
Le guide d'utilisation simple est le suivant.
- Cliquez sur "Recherche d'échantillons" dans la barre de menus de la page supérieure de ChemTEHATRE.
- Sélectionnez "Biotic --Mammals --Marine mammals" dans le menu déroulant "Sample Type".
- Sélectionnez le nom scientifique "Neophocaena phocaenoides" de Snameri dans le menu déroulant de "Nom scientifique".
- Cliquez sur le bouton "Rechercher".
- Cliquez sur le bouton "Exporter les échantillons (TSV)" pour exporter la liste d'échantillons et sur le bouton "Exporter les données mesurées (TSV)" pour exporter la liste des valeurs de mesure sous forme de fichiers texte délimités par des tabulations.
- Enregistrez le fichier exporté dans n'importe quel répertoire et utilisez-le pour une analyse ultérieure.
data_file = "measureddata_20190826061813.tsv" #Remplacez la chaîne de caractères saisie dans la variable par le nom de fichier tsv de vos données mesurées
chem = pd.read_csv(data_file, delimiter="\t")
chem = chem.drop(["ProjectID", "ScientificName", "RegisterDate", "UpdateDate"], axis=1) #Supprimer les colonnes en double lors de la jonction avec des échantillons ultérieurement
sample_file = "samples_20190826061810.tsv" #Remplacez la chaîne de caractères entrée dans la variable par le nom de fichier tsv de vos échantillons
sample = pd.read_csv(sample_file, delimiter="\t")
Le fichier TSV téléchargé est lu par la fonction pandas (read_csv). À ce stade, il est nécessaire de changer le chemin de chaque fichier ("data_file" et "sample_file" dans la cellule supérieure) en un nom de fichier adapté à chaque environnement. L'exemple de code est écrit dans un environnement où le fichier de données se trouve dans le même répertoire que le bloc-notes.
## Chap.3 Préparation des données
Les données traitées cette fois-ci sont une compilation des résultats de mesure de diverses substances chimiques. De plus, les résultats de mesure et les données d'échantillon sont dispersés. En d'autres termes, il est difficile à manipuler tel quel, il est donc nécessaire de le façonner en une forme facile à manipuler. Plus précisément, les opérations suivantes sont nécessaires.
Combinez l'échantillon avec - chem.
- Extraire uniquement les données de mesure des substances chimiques manipulées cette fois.
- Supprimez les colonnes qui ne sont pas nécessaires pour la visualisation.
Sec.3-1 Extraction des données nécessaires
Tout d'abord, procédez aux étapes 1 et 2 ci-dessus.
La liaison consiste à ajouter une base de données d'échantillon (échantillon) d'un échantillon de chaque substance chimique à la base de données (chim) de chaque substance chimique. Ici, le processus d'attachement des données d'échantillon correspondant à l'ID d'échantillon de chaque donnée de chim sur le côté droit de la chimie est effectué.
df = pd.merge(chem, sample, on="SampleID")
Dans les données traitées cette fois, chaque substance chimique est classée de manière assez détaillée. Vous pouvez visualiser les résultats de mesure pour chaque substance chimique, mais cette fois, nous voulons visualiser le contour des résultats de mesure afin que vous puissiez voir en un coup d'œil, nous allons donc extraire les données de la valeur totale de chaque groupe lorsque les substances chimiques sont grossièrement classées. ..
De plus, les données traitées cette fois-ci contiennent un mélange de deux unités différentes. Comme il est inutile de comparer différentes unités, cette fois, nous diviserons les données pour chaque unité. Dans le code ci-dessous, l'unité est spécifiée dans la première ligne de chacun, et celles converties en lipides et humides sont extraites, puis seules les variables commençant par Σ sont extraites. Dans la deuxième ligne, les variables qui commencent par Σ mais ne représentent pas la valeur totale des concentrations de substances chimiques sont en fait supprimées des données.
data_lipid = df[(df["Unit"] == "ng/g lipid") & df["ChemicalName"].str.startswith("Σ")]
data_lipid = data_lipid[(data_lipid["ChemicalName"] != "ΣOH-penta-PCB") & (data_lipid["ChemicalName"] != "ΣOH-hexa-PCB")
& (data_lipid["ChemicalName"] != "ΣOH-hepta-PCB") & (data_lipid["ChemicalName"] != "ΣOH-octa-PCB")]
data_wet = df[(df["Unit"] == "ng/g wet") & df["ChemicalName"].str.startswith("Σ")]
data_wet = data_wet[(data_wet["ChemicalName"] != "ΣOH-penta-PCB") & (data_wet["ChemicalName"] != "ΣOH-hexa-PCB")
& (data_wet["ChemicalName"] != "ΣOH-hepta-PCB") & (data_wet["ChemicalName"] != "ΣOH-octa-PCB")]
Aperçu des données
C'est le résultat du traitement avec Sec.3-1 (uniquement pour ceux dont l'unité est ng / g lipide). En supprimant les données inutiles, seules 131 données pour 6 substances chimiques ont pu être extraites.
```python
data_lipid
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
AlternativeData |
Unit |
Remarks_x |
ProjectID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4372 |
24768 |
SAA001903 |
CH0000096 |
ΣPCBs |
EXA000001 |
5700.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4381 |
24754 |
SAA001903 |
CH0000138 |
ΣPBDEs |
EXA000001 |
170.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4385 |
24902 |
SAA001903 |
CH0000142 |
ΣHBCDs |
EXA000001 |
5.6 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4389 |
24810 |
SAA001903 |
CH0000146 |
ΣHCHs |
EXA000001 |
1100.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| ... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
| 4812 |
25125 |
SAA001940 |
CH0000152 |
ΣCHLs |
EXA000001 |
160.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4816 |
25112 |
SAA001941 |
CH0000033 |
ΣDDTs |
EXA000001 |
9400.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4817 |
25098 |
SAA001941 |
CH0000096 |
ΣPCBs |
EXA000001 |
1100.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4818 |
25140 |
SAA001941 |
CH0000146 |
ΣHCHs |
EXA000001 |
41.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4819 |
25126 |
SAA001941 |
CH0000152 |
ΣCHLs |
EXA000001 |
290.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
131 rows × 74 columns
Sec.3-2 Supprimer les colonnes inutiles
Ensuite, supprimez les colonnes inutiles. En général, si la quantité d'informations est la même, il est préférable de traiter une petite quantité de données, il est donc préférable de supprimer les données inutiles. Dans la trame de données traitée cette fois, il y a de nombreux espaces sans données. Ceci est traité comme N / A (nombre à virgule flottante) sur python, et bien qu'il n'y ait pas d'informations, cela consomme une certaine quantité d'espace. Par conséquent, toutes les colonnes N / A (colonnes sans informations) sont supprimées du bloc de données.
```python
data_lipid = data_lipid.dropna(how='all', axis=1)
data_wet = data_wet.dropna(how='all', axis=1)
```
Aperçu des données
Ceci est le résultat du traitement de Sec.3-2. On peut voir que le nombre de colonnes (la valeur des colonnes apparaissant en bas du tableau) est considérablement plus petit que le résultat de la section 3-1.
```python
data_lipid
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
Unit |
ProjectID |
SampleType |
TaxonomyID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4372 |
24768 |
SAA001903 |
CH0000096 |
ΣPCBs |
EXA000001 |
5700.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4381 |
24754 |
SAA001903 |
CH0000138 |
ΣPBDEs |
EXA000001 |
170.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4385 |
24902 |
SAA001903 |
CH0000142 |
ΣHBCDs |
EXA000001 |
5.6 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4389 |
24810 |
SAA001903 |
CH0000146 |
ΣHCHs |
EXA000001 |
1100.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| ... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
| 4812 |
25125 |
SAA001940 |
CH0000152 |
ΣCHLs |
EXA000001 |
160.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4816 |
25112 |
SAA001941 |
CH0000033 |
ΣDDTs |
EXA000001 |
9400.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4817 |
25098 |
SAA001941 |
CH0000096 |
ΣPCBs |
EXA000001 |
1100.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4818 |
25140 |
SAA001941 |
CH0000146 |
ΣHCHs |
EXA000001 |
41.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4819 |
25126 |
SAA001941 |
CH0000152 |
ΣCHLs |
EXA000001 |
290.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
131 rows × 37 columns
Visualisation des ΣDDT Chap.4
En traitant dans
Chap.3, nous avons préparé des données pour chacun des deux types de substances chimiques. Ici, tout d'abord, nous allons visualiser comment les données sont dispersées sur les ng / g lipides des ΣDDT. </ p>
Sec.4-1 Préparation des données
Tout d'abord, pour la visualisation, les données ΣDDT sont extraites de data_lipid.
```python
data_ddt = data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"]
ddt_vals = data_ddt.loc[:, "MeasuredValue"]
data_ddt
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
Unit |
ProjectID |
SampleType |
TaxonomyID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4401 |
24783 |
SAA001904 |
CH0000033 |
ΣDDTs |
EXA000001 |
140000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4431 |
24784 |
SAA001905 |
CH0000033 |
ΣDDTs |
EXA000001 |
140000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4461 |
24785 |
SAA001906 |
CH0000033 |
ΣDDTs |
EXA000001 |
130000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4491 |
24786 |
SAA001907 |
CH0000033 |
ΣDDTs |
EXA000001 |
280000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
5 rows × 37 columns
Sec.4-2 Statistiques de base
Comme les données ont été extraites à la Sec.4-1, les statistiques de base sont calculées afin de saisir les caractéristiques de cet ensemble de données. Les statistiques de base sont des statistiques qui résument l'ensemble de données. Cette fois, le nombre total de données, la moyenne, l'écart type, le premier quadrant, la valeur médiane, le troisième quadrant, la valeur minimale et la valeur maximale sont calculés.
```python
count = ddt_vals.count()
mean = ddt_vals.mean()
std = ddt_vals.std()
q1 = ddt_vals.quantile(0.25)
med = ddt_vals.median()
q3 = ddt_vals.quantile(0.75)
min = ddt_vals.min()
max = ddt_vals.max()
count, mean, std, q1, med, q3, min, max
```
(24, 102137.5, 81917.43357743236, 41750.0, 70500.0, 140000.0, 9400.0, 280000.0)
Dans la cellule supérieure, les statistiques de base sont calculées individuellement, mais pandas a une fonction qui génère les statistiques de base ensemble.
avgs = ddt_vals.describe()
avgs
count 24.000000
mean 102137.500000
std 81917.433577
min 9400.000000
25% 41750.000000
50% 70500.000000
75% 140000.000000
max 280000.000000
Name: MeasuredValue, dtype: float64
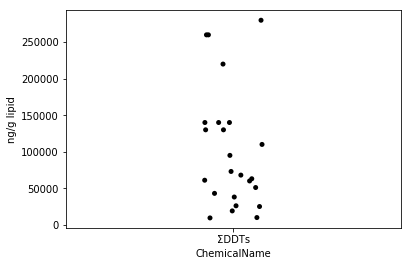
Sec.4-3 jitter plot
Pour visualiser la dispersion des données, il existe un diagramme de dispersion qui trace toutes les données (Eguchi: est-ce un diagramme de gigue, pas un diagramme de dispersion?). En Python, vous pouvez facilement dessiner un tracé de gigue en utilisant la fonction stripplot de seaborn. Le tracé de gigue est l'une des bonnes méthodes de visualisation qui peuvent facilement décrire chaque paradis en ajoutant des fluctuations aux points même lorsque les points se chevauchent.
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_ddt, color='black', ax=ax) #Dessin de tracé de gigue
ax.set_ylabel("ng/g lipid")
plt.show()
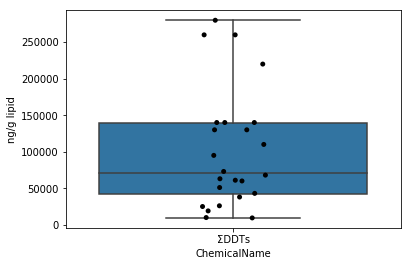
Sec.4-4 Moustaches de boîte
Contrairement aux Sec.4-3, le diagramme de moustaches de boîte trace un résumé de l'ensemble de données avec des statistiques de base. Les moustaches de boîte peuvent être dessinées avec la fonction boxplot de seaborn. Ici, en écrasant le diagramme des moustaches sur le diagramme de gigue précédent, un diagramme qui facilite la compréhension de la distribution des données est généré.
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"], color='black', ax=ax) #Dessin de tracé de gigue
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"], ax=ax) #Dessiner une boîte de moustaches
ax.set_ylabel("ng/g lipid")
plt.show()
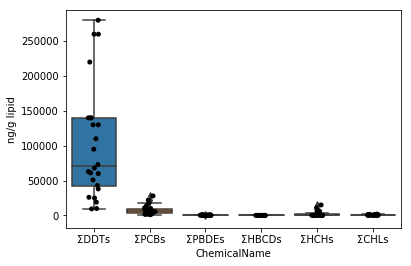
Visualisation des données de chaque substance chimique du chapitre 5 Snameri ( Neophocaena phocaenoides </ i>)
Dans le Chap.4, seule la valeur des ΣDDT a été visualisée. Appliquez-le pour visualiser les données data_lipid et data_wet.
### Section 5-1 Moustaches en boîte (application de l'article 4-4)
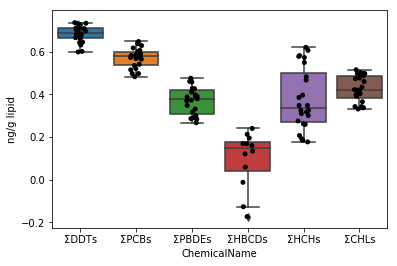
Commencez par dessiner un graphique pour les données de data_lipid de la même manière que dans les Sec.4-4. Puisque la fonction stripplot / boxplot a une fonction pour classer automatiquement les données par le nom de colonne affecté au paramètre x, il suffit d'affecter les données collectées aux données de paramètres telles quelles.
```python
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax) #Dessin de tracé de gigue
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax) #Dessiner une boîte de moustaches
ax.set_ylabel("ng/g lipid")
plt.show()
```
Sec.5-2 Amélioration de la Sec.5-1
Dans le graphique ci-dessus, l'espacement sur l'axe des y est large car les ΣDDT varient considérablement. Par conséquent, les 5 types de graphiques restants sont écrasés et difficiles à voir. Par conséquent, le graphique est rendu plus facile à voir en prenant la valeur logarithmique des données.
Premièrement, lors de la prise du logarithme, le logarithme de 0 ne peut pas être pris, donc vérifiez si l'ensemble de données contient 0. En utilisant l'instruction in, la présence ou l'absence de la valeur correspondante est renvoyée dans TRUE / FALSE.
0 in data_lipid.loc[:, "MeasuredValue"].values
False
Puisqu'il a été trouvé dans la cellule supérieure que l'ensemble de données ne contient pas 0, la valeur logarithmique peut être considérée comme telle.
```python
data_lipid.loc[:, "MeasuredValue"] = data_lipid.loc[:, "MeasuredValue"].apply(np.log10)
```
C:\Users\masah\Anaconda3\lib\site-packages\pandas\core\indexing.py:543: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: http://pandas.pydata.org/pandas-docs/stable/indexing.html#indexing-view-versus-copy
self.obj[item] = s
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax) #Dessin de tracé de gigue
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax) #Dessiner une boîte de moustaches
ax.set_ylabel("log(ng/g) lipid")
plt.show()
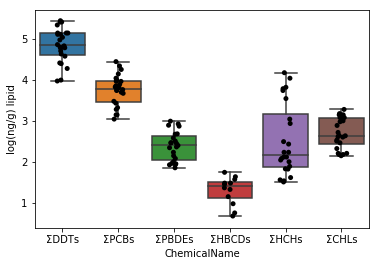
Visualisez
data_wet de la même manière. Premièrement, si l'ensemble de données contient 0, remplacez plutôt 1/2 de la valeur minimale. Après cela, le logarithme est pris et visualisé. </ p>
if 0 in data_wet.loc[:, "MeasuredValue"].values:
data_wet["MeasuredValue"].replace(0, data_wet[data_wet["MeasuredValue"] != 0].loc[:, "MeasuredValue"].values.min() / 2) #Valeur minimale 1/Suppléant 2
else:
pass
data_lipid.loc[:, "MeasuredValue"] = data_lipid["MeasuredValue"].apply(np.log10)
data_lipid.loc[:, "Unit"] = "log(ng/g lipid)"
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax)
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax)
ax.set_ylabel("log(ng/g lipid)")
plt.show()
note de bas de page
1 Notez qu'il s'agit d'une fonction Juyter Notebook appelée "commande magique" et ne peut pas être utilisée avec du code python général.
2 Abréviation des valeurs séparées par des tabulations. Il s'agit d'un format de fichier dans lequel les données sont séparées par des caractères TAB (à l'extrémité gauche du clavier). Convient pour contenir des données en forme de tableau. Un format de fichier similaire est CSV (abréviation des valeurs séparées par des virgules, séparées par ", (virgule)").
